

Scientists explore the physiological irony that high blood pressure kills cells, which generates trash that further raises blood pressure.
It’s a vicious and apparently unhealthy circle. It’s a vicious and apparently unhealthy circle. While we’ve known for decades that a bacterial infection can raise our blood pressure short term, now scientists are putting together the pieces of how our own dying cells can also trigger an immune response that fuels chronically high, destructive pressure.
Quite literally billions of our cells die daily, most often as part of natural turnover, their remains mostly taken up by garbage-eating immune cells like macrophages. A burned tongue from a hot cup of morning coffee that is mostly better by the afternoon illustrates normal cell death and typically rapid recovery.
But the die rate increases as we age and so do rates of hypertension.
A waste-management system pushed beyond capacity
High blood pressure increases cell carnage, and scientists have increasing evidence that the body’s waste management system gets overwhelmed like a waste processing plant operating beyond capacity. The resulting mounting debris gets the attention of our immune system, causing inflammation and blood vessel constriction that contributes to worsening hypertension and even more cell death from the pressure.
“It’s a circle,” says Dr. R. Clinton Webb, chairman of the Department of Physiology at the Medical College of Georgia at Augusta University, in which inflammation is a constant and which isn’t addressed in today’s treatment regimens.
“The more blood pressure goes up, the more injury you have,” says Webb, and he and his colleagues want to understand the signaling pathways that are driving this inflammation that further drives blood pressure.
Webb, a vascular expert and Herbert S. Kupperman Chair in Cardiovascular Disease, is principal investigator on a $9.4 million Program Project Grant from the National Heart, Lung and Blood Institute, which will further parse how contents regularly spilt from a surplus of dying cells contribute to this circle.
While the chicken or the egg question isn’t answered, it’s clear that higher blood pressure – from causes like obesity to genetics to salt sensitivity – increases cell death rates throughout the body. Once outside the dying cell, typical contents like DNA as well as HMGB1, which helps stabilize the DNA, are categorized as damage-associated molecular patterns, or DAMPs.
The scientists call DAMPs “alarm signals” because they look like bacterial or viral invaders to the immune system. That sets in motion likely well-intended inflammation to fight the invader, but blood-vessel constriction and narrowing, kidney and other organ damage, and more cell death also follow.
The new grant is enabling Webb and his team to look at issues like what happens when the increased carnage inside blood vessel walls increases the release of DNA from cell powerhouses called mitochondria – DNA that actually is distinctly different from our own.
Project leaders Drs. Jennifer Sullivan and Paul O’Connor, respectively, are exploring how the different ways cells die in males and females impact the damage that follows the release of HMGB1 and how an onslaught of these and other DAMPs cripple the kidney’s ability to help regulate blood pressure.
Dr. Adviye Ergul, vascular physiologist, is managing the bioinflammation core, and Dr. Michael Brands, cardiovascular-renal physiologist, is managing the animal use and instrumentation core for the five-year studies to ensure the consistency of results gathered and minimal animal usage.
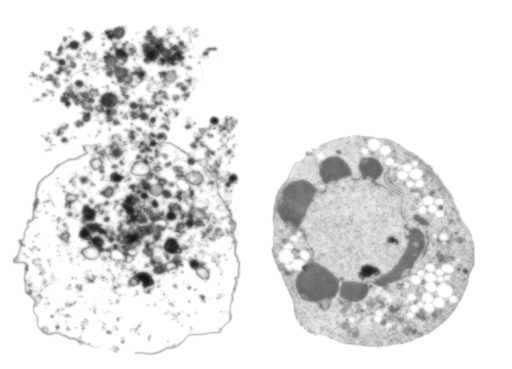
A cause and effect
Hypertension affects about one-third of the adult population in the United States, according to the Centers for Disease Control and Prevention. About 85 percent of the time, the true cause is unclear. Webb and his colleagues think DAMPs are both the result of and initiators of hypertension in many.
“Cells are dying all over if you have hypertension,” Sullivan says. Smooth muscle cells and endothelial cells that comprise the blood vessels are definite casualties. “There is no doubt there are more dead cells in the hypertensive blood vessel,” Webb says.
All this internal debris has to go somewhere, and some finds its way into nearby cells. There it can activate immune receptors that, instead of just consuming the debris, mount a fight that further damages the form and function of blood vessels and organs, Webb says.
There is no doubt there are more dead cells in the hypertensive blood vessel.
– Dr. Clinton Webb
Like bacteria, DAMPs activate Toll-like receptors, or TLRs, that can be found inside many cells and outside others, although they are known to translocate. The nine different types of TLRs also have in common some degree of activation of MYD88, a downstream molecule, which in turn activates transcription factor NF-kB, which helps control gene expression as well as cell death and growth. In the case of activation by DAMPs, TLRs initiate inflammation, a good reaction when, for example, TLR4 responds as it should to a membrane component of bacteria.
Webb’s team is focusing on what happens inside blood vessels when excessive cell death leads to excessive release of the mitochondrial DNA. Even with its foreign DNA, as with other DAMPs, all is fine until mitochondrial DNA gets spilled. At that moment, it can start to activate TLR9s inside immune, vascular smooth muscle or endothelial cells, Webb says. One of the many questions the researchers want to answer is whether TLR4 and TLR9 have some unfortunate inflammatory synergy through their activation of MYD88 in this scenario.
The long-term goal of Webb and his colleagues is identifying targets that could block some of the unintended results without putting people at risk for additional disease by generally suppressing their natural immune response.
“Imagine, for example, if you could figure out a drug that would actually block say the uptake of mitochondrial DNA,” Webb says. Or, in the case of Sullivan, a pharmacologist and physiologist, a drug that reduces excessive activation of T cells, the drivers of the immune response.
“There is this growing interest in the adaptive immune response [which colleague and cancer immunologist Dr. Kebin Liu likens to a targeted missile strike] and T cells being important in causing increases in hypertension,” Sullivan says of a finding that has been shown in animal models and people. “There is evidence of T-cell activation, but what is causing it?” Knowing that answer could lead to treatment that selectively blocks excess activation, so the scientists are looking upstream for activators.
Like most things in the body, there are balances, in this case effector T cells that promote inflammation and Tregs that reduce it. Effector T cells are definitely a contributor to hypertension, and DAMPs are likely one of their activators, particularly in males, Sullivan says.
Cell death in males tends to be messier
In males, high blood pressure tends to kill cells through necrosis, in which cells essentially burst open. At least before the age of menopause, females experience more apoptosis, or programmed cell death — a natural, cleaner approach to cell elimination that better contains their innards.
“The way a cell dies is important, and the males are releasing things to a greater degree,” Sullivan says. On the other hand, apoptosis, in which the cell engulfs itself, is actually anti-inflammatory and appears instead to attract calming Tregs. Sullivan and her team have watched this scenario play out in isolated kidney and heart cells.
“Both sexes are becoming hypertensive, but it doesn’t mean they are taking the same path to get there,” she says. And, if they can learn more about the differences – along with any common ground – they can devise more effective treatment for at least a segment of patients with hypertension.
So the new grant is enabling a further look at how cells die in male versus female hypertensive rats and, when scientists inhibit each cell death method, what that does to blood pressure regulation as well as which T cells get activated. While Sullivan isn’t certain she could or would make the males apoptose, right now she wants to know more about which cells are dying in the kidneys, how they are dying and what immune response gets activated as a result in the males versus females. A future goal may instead be inhibiting necrosis in males.
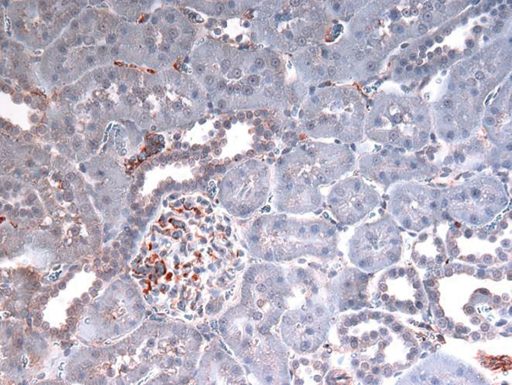
The red/brown staining indicates the presence of HMGB1.
One of the many things necrosis dumps — and that the scientists are following — is the DNA-stabilizing molecule HMGB1. Outside the cell, HMGB1 gets the attention of the immune system, in this case, TLR4. Their theory and early evidence indicates more HMGB1 is dumped in the males than in their female counterparts.
Kidney’s salt sandpit often a target
O’Connor, a renal physiologist, is also focusing on the kidneys. He wants to test the hypothesis that high levels of DAMPs interfere with the critical work of the middle portion of the kidney. This renal medulla, which is directly linked to the tubules that collect urine, is critical to concentrating urine so you can still eliminate salt and other byproducts, particularly when you are not consuming enough liquid.
O’Connor likens the renal medulla to a sandpit for salt, and the unique, long, thin-walled U-shaped blood vessels help ensure that all salt – which the kidneys use to help regulate fluid volume and blood pressure – is not lost.
However, the odd vascular configuration that O’Connor calls an “Achilles’ heel” also makes the blood vessels susceptible to clogging. “We think they are always susceptible to getting clogged up. The red blood cells have a tendency to get trapped in there,” he says.
There appears to be a sort of traffic circle to help keep things moving because these blood vessels are lined by tiny contractile cells called pericytes that O’Connor calls “24/7 plumbers on demand.”
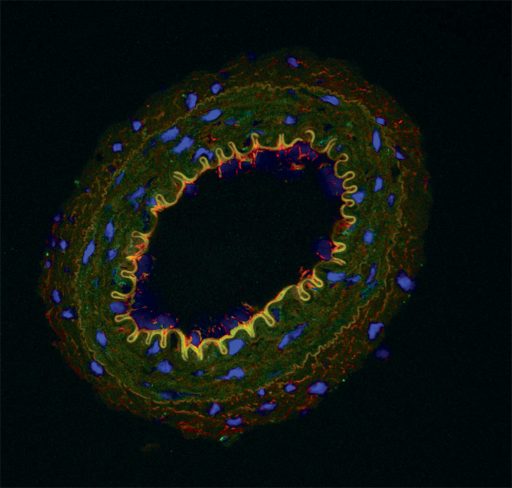
“We think DAMPs might hamper this contraction and allow red blood cells to get stuck. As that slowly happens, it changes the kidney function and contributes to high blood pressure,” O’Connor says. They know this happens with the odd-shaped red blood cells of sickle cell disease. Their new pursuits include exploring whether endothelial cells that line the unusual vasculature are stimulated by DAMPs to make more of the blood vessel dilator nitric oxide, which weakens the contractility of pericytes.
We think DAMPs might hamper this contraction and allow red blood cells to get stuck. As that slowly happens, it changes the kidney function and contributes to high blood pressure.
– Dr. Paul O’Connor
If the theory and early evidence hold, drugs that support the contractile cells could enable the kidneys to remain helpful in avoiding hypertension, rather than worsening it. The researchers note the vast capacity of the kidneys to support us, and that related problems typically do not become apparent until about 80 percent of function is lost.
“We are trying to identify whether DAMPs play a role in the hypertensive process. I think that is the fundamental question,” Webb says of the extensive studies ahead.
Hypertension affects about one-third of the adult population in the United States, according to the Centers for Disease Control and Prevention. About 85 percent of the time, the true cause is unclear.
Cancer Connection
In a related study recently added to the already long list, Dr. R. Clinton Webb, chairman of the MCG Department of Physiology, is collaborating with cancer immunologist Dr. Kebin Liu, professor in the MCG Department of Biochemistry and Molecular Biology and at the Georgia Cancer Center, to explore the fact that hypertension also is emerging as the most common comorbidity – which is collateral damage essentially – of chemotherapy. Like high blood pressure, cancer drugs increase cell death, and resulting cellular debris, including DAMPs, again appears to catch the attention of the immune system. Cancer patients may later find themselves with hypertension and other cardiovascular damage. So Webb and Liu are giving rats tumors, treating them with chemotherapy and then looking at what happens to their blood vessels to further clarify the association. Liu wants to do parallel studies on the effects of immunotherapy, which is increasingly packaged with chemotherapy. Considered a bit of a more natural approach than chemotherapy, immunotherapy turns to the body’s own medicine cabinet to better place cancer in the bull’s-eye of natural defense processes. As an example, with adoptive T-cell therapy, patients’ T cells — which drive their immune response — are removed, reengineered to better fight their cancer, then returned to them. The new and perhaps unusual collaborators note that cancer and cardiovascular problems such as hypertension do share some risk factors like smoking and obesity. They also share inflammation. Liu and Webb hope their joint studies will determine whether increased cellular trash is additional common ground.








