

A Critical Junction
Dr. Ferenc “Frank” Deak describes a chemical synapse as “really where all the action happens in the brain,” a point where an action potential, basically an electrical message from the neuron, triggers the release of a chemical message, or neurotransmitter.
Aging is the biggest known risk factor for Alzheimer’s, according to the Alzheimer’s Association, and the buildup of beta amyloid plaque, a hallmark of the disease.
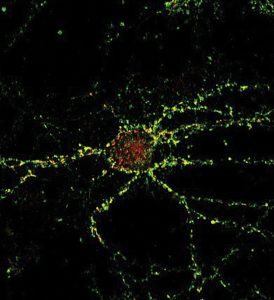
In what feels like a twisted tale, neuroscientist Deak studies how the synaptic connections influence the buildup that ultimately destroys them.
Beta amyloid is a piece of amyloid precursor protein, one of the highest expressed proteins in a healthy brain. The protein is essential to the developing brain and apparently to the developed brain as well, including providing all sorts of signaling functions, although exactly what remains unclear, Deak says.
However it does appear that to do its jobs, amyloid precursor protein needs to be cut into smaller sections, and under the wrong circumstances, one of the pieces cut out is sticky beta amyloid.
The sticky pile can build over maybe 20 years or more, until it becomes a substantial plaque outside neurons that interferes with their ability to communicate. The pile also triggers destructive inflammation. As damage progresses outside, inside the neurons another twisted collection of the protein tau, called neurofibrillary tangles, coincides with the onset of dementia.
“Connections get lost between neurons,” says Deak, and by then it’s too late. “We want to do something before that.”
The way communication is supposed to happen is that with the help of a family of proteins called SNARE, a biological carrying case called a vesicle fuses to the membrane of the presynaptic neuron — the neuron sending the message. That creates an opening in the cell membrane at its outer edge and near the receiving end of the postsynaptic neuron, so the neurotransmitter and other contents of the vesicle can be released, in a process called exocytosis. Then in rapid succession, the empty vesicle essentially collapses into the membrane of the presynaptic neuron so it can be taken back inside the neuron, retooled and reused.
But like the rest of our body, vesicles change with age and with Alzheimer’s.
Untwisting the tale
In a bit of a perfect storm, synaptic vesicles in the older brain tend to pick up more amyloid precursor protein from the membrane of the neuron during their refresh. When the flattened biological carrying case gets brought back inside the neuron, the increased amount of amyloid precursor protein it brings with it can encounter enzymes called beta and gamma secretases, which process membrane proteins. But in this case, they are problematic, excising the sticky beta amyloid. Inside the neuron, the sticky neurotoxic amyloid starts making bad changes, including impacting the vesicles.
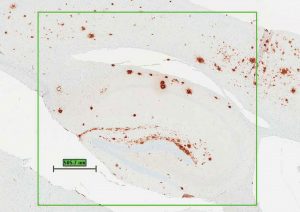
brown spots are destructive plaques.
Later other vesicle types will start hauling it out, dumping the neurotoxin at the synapse. One unfortunate failure in Alzheimer’s treatment was drugs that tried to improve the scenario by inhibiting the secretases that would make two cuts instead of one, so beta amyloid would not be freed, but there were odd consequences like bleeding from the gut and turning hair permanently orange.
Deak has mounting evidence that a protein on the surface of the vesicle’s membrane, might be a better point of action.
He collaborated with the Mayo Clinic Brain Bank to look in diseased and healthy brains for genetic variants, including synaptic protein genes, which might be players in Alzheimer’s.
He found that with age the amount of synaptobrevin-1, or VAMP1, increases in the membrane, or surface, of the vesicle. As the name implies, synaptobrevin-1 is a usual constituent of synaptic vesicles, aiding their fundamental function of neurotransmitter release.
Single nucleotide polymorphisms, or SNPs, are comparatively simple genetic variations and the most common variations found in humans; and genetic screens of humans have found SNPs for synaptobrevin-1 are “significantly” associated with late onset Alzheimer’s disease. Also, SNPs associated with increased synaptobrevin-1 expression increase the risk of Alzheimer’s, and those that reduce its expression appear to reduce the risk. And, in synaptobrevin-1 knockout mice, beta amyloid levels are significantly reduced. Deak and his colleagues hypothesize that variations in the synaptobrevin-1 level alter the relationship between synaptic transmission and the processing of amyloid precursor protein and consequently have a profound effect on beta amyloid production. A bottom line is that high synaptobrevin-1 levels mean amyloid precursor protein is cleaved in a way that produces more of that neurotoxic amyloid. With age, all of us collect some amyloid plaque as well as neurofibrillary tangles, but in Alzheimer’s there is so much that it interferes with neuron function. Deak thinks the vesicle’s role in the destructive tale is an early one that a $1.9 million grant from the National Institute on Aging is helping him further explore.
Right now he and his research team are looking at the impact of synaptobrevin-1 levels on the activity of synapses and production of beta amyloid in their knockout mice, and what reducing synaptobrevin-1 levels does to reduce cognitive impairment in a mouse model of Alzheimer’s. They hypothesize that lower synaptobrevin-1 will reduce the unhealthy association between the level of synaptic activity, how amyloid precursor protein gets processed and how much neurotoxic amyloid ultimately gets dumped on synapses.
If they are correct, synaptobrevin-1 may emerge as an important regulator of amyloid production in the synapse and a novel target for diagnosing and treating Alzheimer’s.

Back Story
Deak was born in Hungary, a country about the size of Indiana in Central Europe. He earned a doctor of medicine degree, summacum laude, from Semmelweis University of Medicine in Budapest, in 1994, and a PhD in the university’s Department of Physiology, also summa cum laude, in 2000. To support himself during graduate school, Deak was also a faculty member and lecturer in the Physiology Department, and on the weekends he pulled long shifts on ambulances, delivering babies, working accident scenes and making house calls. He came to the United States right after grad school, to complete postdoctoral studies with Dr. Thomas C. Südhof in the Center for Basic Neuroscience at the University of Texas Southwestern in Dallas. Five years later, Südhof would win a Nobel Prize for his work to understand the machinery regulating vesicle traffic, including how they stay in place, ready to release signals at the right moment with the help of calcium sensing proteins. Deak had originally loved the idea ofmbeing a neurologist — which made him a bit of the odd man out in a family focused on engineering — but decided that by being a neuroscientist he could discover more about the fundamentals that go wrong in the brain, like problems with those vesicles.
So next, Deak joined the faculty of the Department of Neuroscience at the Mayo Clinic in Jacksonville to focus on Alzheimer’s research. In 2012, he joined the faculty of the Reynolds Oklahoma Center of Aging and Oklahoma Center for Neuroscience at the University of Oklahoma Health Sciences Center in Oklahoma City and came to MCG late last year. Over the years, he has served as a member of numerous NIH study sections, including the Chronic Dysfunction and Integrative Neurodegeneration Study Section. His work has been published in top journals like Science, Neuron and Nature Cell Biology. He is associate editor of Geroscience, the journal of the American Aging Association. The word geroscience is defined by the National Institute on Aging as the intersection of basic aging biology, chronic disease and health. As his long-term goal, Deak aims to use geroscience approaches to prevent multiple chronic brain disorders of the elderly.








